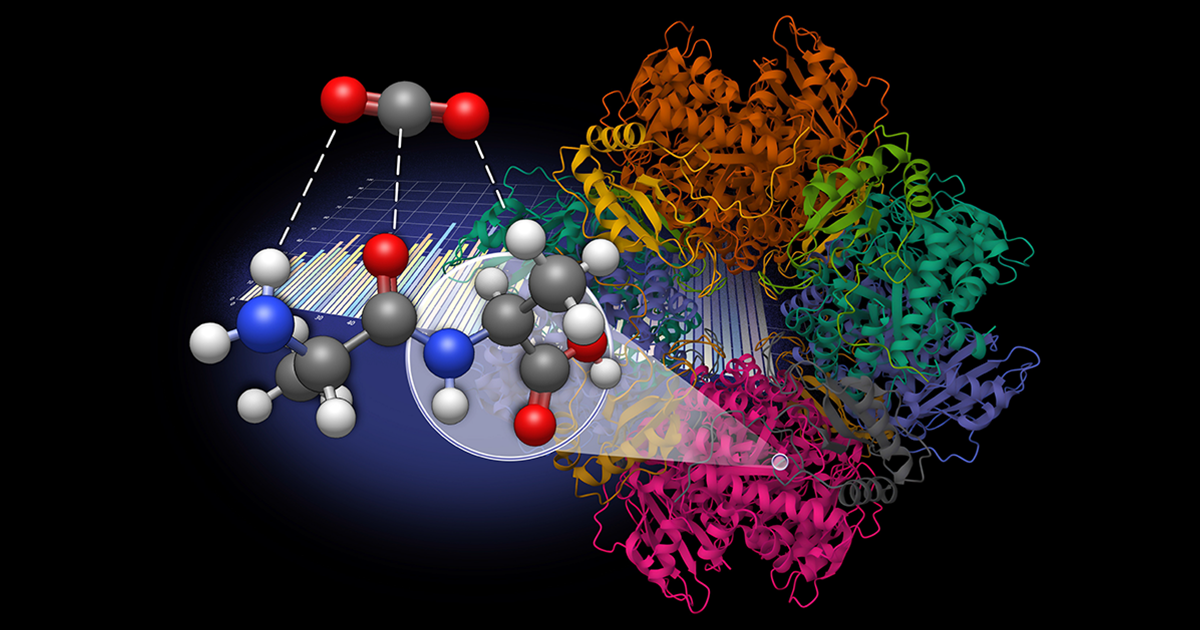
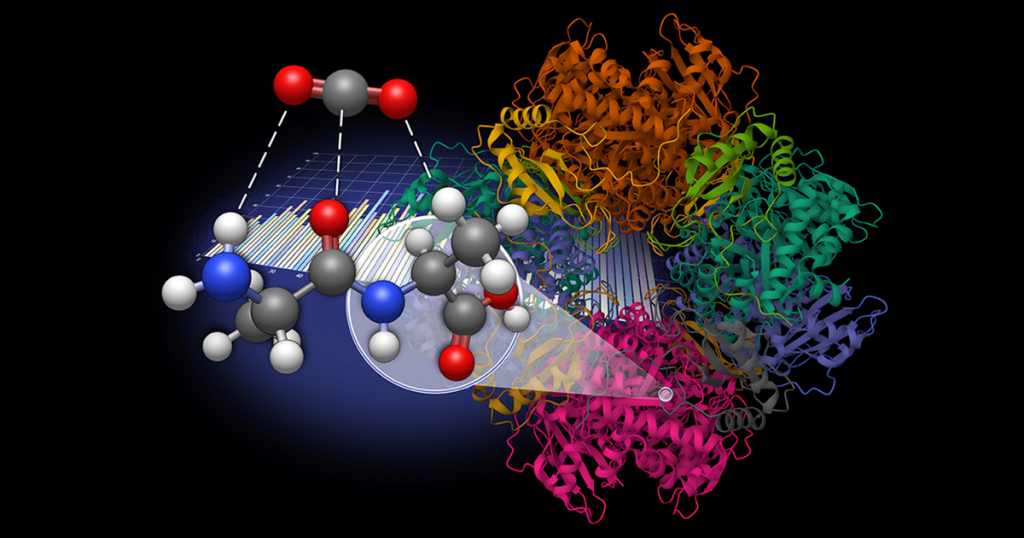
UT Chemistry Lab Explores Dipeptides for Carbon Dioxide Capture
Associate Professor Konstantinos Vogiatzis’ lab in the Department of Chemistry is leveraging computational chemistry to address excess carbon dioxide (CO2) in the atmosphere.
The presence of excess CO2 in the atmosphere is believed to have a number of far-reaching impacts on the environment. Over the last 60 years the amount of CO2 in the atmosphere has more than tripled. Today, carbon dioxide levels are estimated to be higher than ever before in human history. The presence of such high levels of CO2 in the atmosphere is believed to have a number of far-reaching impacts on the environment.
One common method of managing excess CO2 is carbon capture and storage (CCS). CCS usually employs amine-based solvents to trap CO2 and prevent it from moving into the atmosphere. However, this method has some limitations. The solvents used in this process are expensive, volatile, and can produce harmful byproducts that may increase cancer risks in humans.
Seeking a more sustainable solution, Vogiatzis, graduate student Amarachi Sylvanus, and post-doctoral researcher Grier Jones explored dipeptides as a natural, bioinspired alternative for CO2 sequestration. This work was done in collaboration with Radu Custelcean, distinguished research scientist at Oak Ridge National Laboratory.
The research team generated a database of 960 dipeptide molecules derived from 20 natural amino acids and developed an automated workflow to model molecular interactions with CO2.
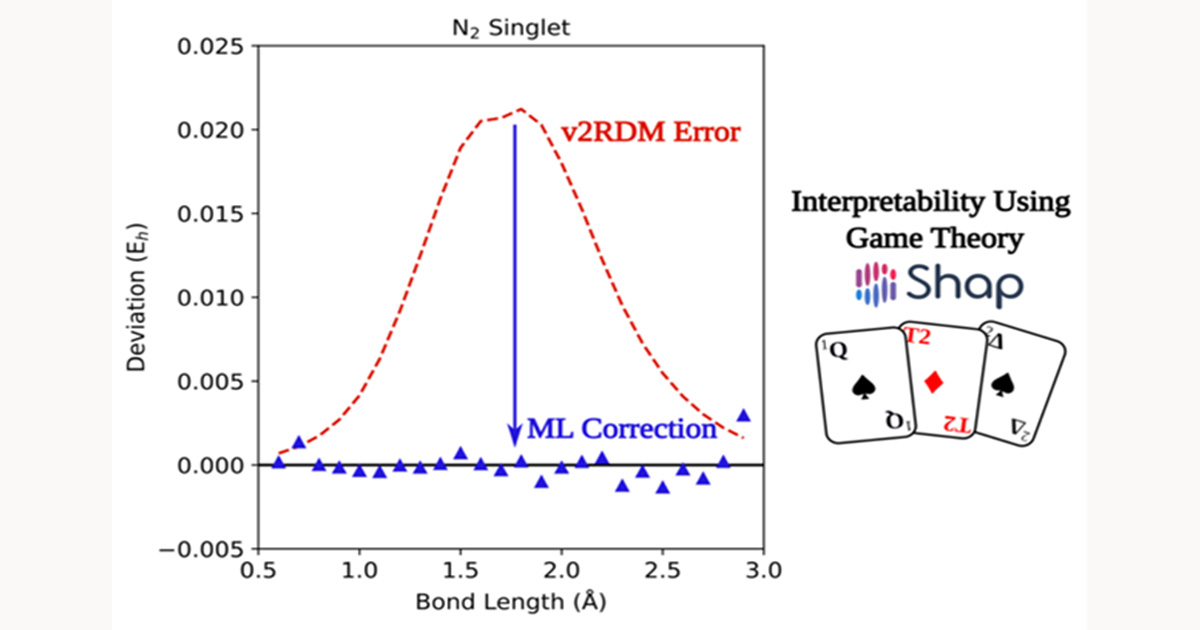
By leveraging density functional theory (DFT) and symmetry-adapted perturbation theory (SAPT), they systematically evaluated interactions between the dipeptides and CO2. Their analysis identified key amino acid subunits that enhance CO2 binding through cooperative effects.
“Our results confirm that cooperative interactions between CO2-philic groups in dipeptides significantly enhance CO2 capture compared to individual amino acids,” said Vogiatzis. “This discovery provides valuable design principles for optimizing CO2 capture efficiency.”
The study revealed that dipeptides exhibit greater interaction energy diversity than their individual amino acid components, highlighting the critical role of cooperative effects. Statistical analysis showed that asparagine subunits frequently strengthen CO2 binding, while glycine subunits tend to weaken it.
Beyond fundamental insights, this research lays the groundwork for industrial applications, particularly in direct air capture (DAC) technologies. DAC is a promising technology that pulls CO2 from air at both concentrated and dispersed locations. By understanding how dipeptides interact with CO2, researchers can guide the development of next-generation carbon capture materials.
“We believe our findings will contribute to the future design of bioengineered materials for large-scale CO2 capture. Nature provides incredible solutions, and by mimicking its mechanisms, we can develop transformative technologies to combat climate change,” said Vogiatzis.
This pioneering study exemplifies the power of computational chemistry and bioinspired design in addressing global environmental challenges.
The results of this study were published in the journal ChemPhysChem and highlighted in ChemistryViews.